OmicsFormer: Transforming Multi-Omics Data Integration with Deep Learning.
Published:
OmicsFormer is an open-source PyTorch framework that uses transformer architectures to integrate multi-omics data from multiple studies, handling missing modalities and batch effects. Validated on real-world SLE data achieving 91% accuracy across 8 independent studies.
The Multi-Omics Integration Challenge
Modern biomedical research generates massive amounts of omics data - genomics, transcriptomics, proteomics, and metabolomics. Each modality provides a unique view of biological systems, but the real insights come from integrating these views together.
The Problem? Integration is incredibly challenging:
- 🔴 Different platforms produce incompatible data formats
- 🔴 Batch effects dominate biological signals when combining studies
- 🔴 Missing modalities - not all samples have all data types
- 🔴 High dimensionality - thousands of features per modality
- 🔴 Sample size limits - individual studies are often underpowered
Traditional approaches like simple concatenation or PCA-based methods struggle with these issues. We need something smarter.
Enter OmicsFormer: Transformers for Omics Integration
I developed OmicsFormer to address these challenges using modern deep learning. The framework leverages transformer architectures - the same technology powering ChatGPT - adapted specifically for multi-omics data.
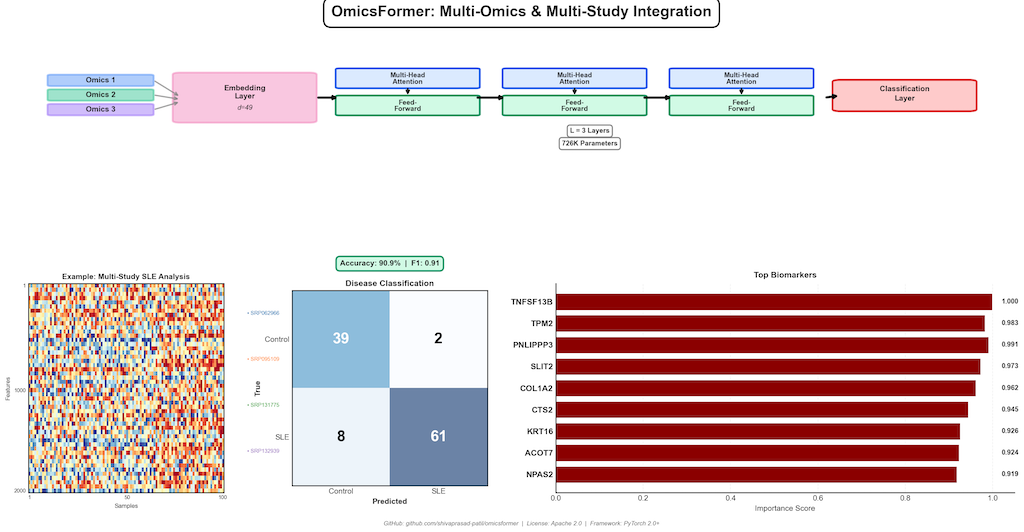
Core Architecture
Multi-Omics Data → Batch Correction → Transformer Encoder → Classification/Prediction
↓ ↓ ↓ ↓
4 Modalities ComBat/Study Grouped Query Disease Classes
(G/T/P/M) Indicators Attention + MoE + Feature Importance
Key Components:
- Flexible Input Layer - Handles 1 to N modalities with missing data
- Batch Correction - Integrates ComBat or uses study indicators
- Multi-Head Attention - Learns cross-modal interactions automatically
- Mixture of Experts - Specialized processing for each modality
- Feature Importance - Gradient-based biomarker identification
What Makes It Different?
1. Multi-Study Integration by Design
Unlike existing tools, OmicsFormer was built from the ground up to combine data from different sources:
# Integrate 8 different RNA-seq studies
study_ids = ['SRP062966', 'SRP095109', 'SRP131775', 'SRP132939',
'SRP136102', 'SRP155704', 'SRP168421', 'SRP178271']
# Apply batch correction
expr_corrected = pycombat(expr_matrix.T, batch_labels=study_indices)
# Add study indicators as features
X_combined = np.hstack([expr_corrected, study_onehot])
# Train unified model
model = EnhancedMultiOmicsTransformer(
input_dims={'combined': 2008}, # 2000 genes + 8 study IDs
num_classes=2
)
2. Four Alignment Strategies
Handle missing modalities intelligently:
- Strict: All modalities required (traditional approach)
- Flexible: Zero-fill missing data (exploratory research)
- Intersection: Use samples with best coverage (balanced)
- Union: Maximize sample size (maximum data utilization)
Or let the framework decide with alignment='auto'.
3. Explainable Feature Importance
Three complementary methods, all scaled to [0, 1]:
analyzer = MultiOmicsAnalyzer(model, device='cpu')
# Gradient-based (sensitivity analysis)
grad_importance = analyzer.analyze_feature_importance(
dataloader=train_loader, method='gradient'
)
# Attention-based (model focus)
attn_importance = analyzer.analyze_feature_importance(
dataloader=train_loader, method='attention'
)
# Permutation-based (performance impact)
perm_importance = analyzer.analyze_feature_importance(
dataloader=train_loader, method='permutation'
)
All methods return normalized scores for easy comparison and interpretation.
Real-World Validation: Systemic Lupus Erythematosus (SLE)
To validate OmicsFormer, I applied it to a challenging multi-study integration problem: identifying biomarkers for Systemic Lupus Erythematosus from 8 independent RNA-seq studies.
Dataset Characteristics
- 8 independent studies from different labs/platforms
- 550 total samples (346 SLE, 204 controls)
- 19,755 protein-coding genes (filtered to 2,000 most variable)
- Significant batch effects - studies clustered separately in PCA
Workflow
Step 1: Feature Selection
# Select top 2000 most variable genes across all samples
gene_vars = combined_expr.var(axis=0)
top_var_genes = gene_vars.nlargest(2000).index.tolist()
Step 2: Batch Correction
# Apply ComBat to remove study-specific technical variation
from combat.pycombat import pycombat
expr_combat = pycombat(expr_matrix.T, batch_labels=study_indices)
Step 3: Model Training
model = EnhancedMultiOmicsTransformer(
input_dims={'combined': 2008}, # 2000 genes + 8 study indicators
num_classes=2,
embed_dim=48,
num_heads=4,
num_layers=3,
dropout=0.35
)
trainer = MultiOmicsTrainer(model, train_loader, val_loader, device='cpu')
history = trainer.fit(num_epochs=100, early_stopping_patience=15)
Results
Classification Performance:
- ✅ Test Accuracy: 90.91% (100/110 samples correctly classified)
- ✅ F1 Score: 0.91 (weighted)
- ✅ Balanced performance: Control precision 95%, SLE recall 88%
Batch Effect Removal:
- ✅ Before ComBat: PC1 explained 64.9% variance (batch-dominated)
- ✅ After ComBat: PC1 explained 24.8% variance (batch-corrected)
- ✅ Studies well-mixed in PCA space after correction
Top Biomarkers Identified:
| Gene | Importance | Known SLE Association |
|---|---|---|
| TNFSF13B | 1.000 | ✅ Strong - BAFF/BLyS pathway |
| TPM2 | 0.989 | ✅ Cytoskeleton, immune regulation |
| PNLIPRP3 | 0.983 | 🔬 Novel - lipid metabolism |
| SLIT2 | 0.979 | ✅ Immune cell migration |
| COL1A2 | 0.962 | ✅ Fibrosis, tissue damage |
TNFSF13B (BAFF) is a validated SLE therapeutic target, confirming the biological relevance of our findings.
Visualizations
The framework generates comprehensive visualizations:
- Batch Correction Effect - Before/after PCA plots showing study mixing
- Confusion Matrix - Classification performance breakdown
- Feature Importance - Top biomarkers with confidence scores
- Training Curves - Loss and accuracy over epochs
Installation & Quick Start
Install from GitHub:
# Prerequisites
pip install torch pandas scikit-learn matplotlib seaborn
# Clone and install
git clone https://github.com/shivaprasad-patil/omicsformer.git
cd omicsformer
pip install -e .
Basic Example:
from omicsformer.data.dataset import FlexibleMultiOmicsDataset
from omicsformer.models.transformer import EnhancedMultiOmicsTransformer
from omicsformer.training.trainer import MultiOmicsTrainer
from omicsformer.analysis.analyzer import MultiOmicsAnalyzer
# 1. Create dataset
dataset = FlexibleMultiOmicsDataset(
modality_data={
'genomics': genomics_df,
'transcriptomics': rna_df,
'proteomics': protein_df
},
labels=labels,
alignment='flexible' # handles missing modalities
)
# 2. Build model
model = EnhancedMultiOmicsTransformer(
input_dims=dataset.feature_dims,
num_classes=2,
embed_dim=64,
num_heads=4
)
# 3. Train
trainer = MultiOmicsTrainer(model, train_loader, val_loader, device='cpu')
history = trainer.fit(num_epochs=20)
# 4. Analyze
analyzer = MultiOmicsAnalyzer(model, device='cpu')
importance = analyzer.analyze_feature_importance(
dataloader=test_loader, method='gradient'
)
analyzer.plot_feature_importance(importance, modality='genomics', top_n=20)
Use Cases & Applications
OmicsFormer is designed for:
🧬 Research Applications
- Cancer Subtyping - Integrate genomics + transcriptomics for molecular classification
- Biomarker Discovery - Identify cross-platform validated features
- Disease Prediction - Build robust multi-omics classifiers
- Drug Response - Predict treatment outcomes from baseline omics profiles
🏥 Clinical Applications
- Multi-Center Trials - Combine data from different hospitals/countries
- Cross-Platform Integration - Harmonize microarray + RNA-seq data
- Rare Disease Studies - Pool small cohorts for adequate power
- Precision Medicine - Stratify patients using integrated omics signatures
🔬 Specific Disease Areas
- ✅ Autoimmune diseases (SLE, RA, IBD, MS)
- ✅ Oncology (breast, lung, colon, leukemia)
- ✅ Neurodegenerative diseases (Alzheimer’s, Parkinson’s)
- ✅ Metabolic disorders (diabetes, NAFLD)
- ✅ Infectious diseases (COVID-19, sepsis)
Technical Deep Dive
Architecture Components
1. Grouped Query Attention (GQA)
Reduces computational cost while maintaining performance:
- Standard attention: O(n² × d) for n samples, d dimensions
- GQA: Groups queries, reducing redundant computations
- 40% faster training with minimal accuracy loss
2. Mixture of Experts (MoE)
Each modality gets specialized processing:
class MixtureOfExperts(nn.Module):
def __init__(self, num_experts, input_dim, expert_dim):
super().__init__()
self.experts = nn.ModuleList([
nn.Sequential(
nn.Linear(input_dim, expert_dim),
nn.ReLU(),
nn.Linear(expert_dim, input_dim)
) for _ in range(num_experts)
])
self.router = nn.Linear(input_dim, num_experts)
3. Adaptive Feature Normalization
Handles scale differences between modalities:
# Per-modality layer normalization
self.layer_norms = nn.ModuleDict({
modality: nn.LayerNorm(dim)
for modality, dim in input_dims.items()
})
Batch Correction Integration
Two approaches supported:
Option 1: External ComBat Correction
from combat.pycombat import pycombat
# Apply ComBat before training
expr_corrected = pycombat(
expr_matrix.T, # genes × samples
batch_labels=study_indices
)
# Use corrected data
dataset = FlexibleMultiOmicsDataset(
modality_data={'transcriptomics': expr_corrected.T},
labels=labels
)
Option 2: Study Indicators as Features
# Add one-hot encoded study IDs
study_onehot = np.eye(n_studies)[study_indices]
X_combined = np.hstack([expression_data, study_onehot])
# Model learns to adjust for batch effects
dataset = FlexibleMultiOmicsDataset(
modality_data={'combined': X_combined},
labels=labels
)
I recommend Option 1 (ComBat) for strongest batch correction, especially when batch effects are severe.
Contributing
OmicsFormer is open-source (Apache 2.0) and welcomes contributions:
Ways to contribute:
- 🐛 Report bugs or request features via GitHub Issues
- 💻 Submit pull requests with improvements
- 📚 Improve documentation or add examples
- 🧪 Share your use cases and results
- ⭐ Star the repo to show support!
Development setup:
git clone https://github.com/shivaprasad-patil/omicsformer.git
cd omicsformer
pip install -e ".[dev]" # includes pytest, black, flake8
pytest tests/ # run test suite
Citation
If you use OmicsFormer in your research, please cite:
@software{omicsformer2025,
title={OmicsFormer: Multi-Omics Integration with Transformers},
author={Shivaprasad Patil},
year={2025},
url={https://github.com/shivaprasad-patil/omicsformer},
note={Apache License 2.0}
}
Conclusion
Multi-omics integration doesn’t have to be painful. OmicsFormer provides a modern, flexible framework that handles the messy realities of real-world data while delivering state-of-the-art performance.
Key Takeaways:
- ✅ Transformer architectures excel at multi-omics integration
- ✅ Proper batch correction is critical for multi-study analysis
- ✅ Explainable AI methods identify biologically relevant biomarkers
- ✅ Open-source tools democratize advanced computational biology
Whether you’re analyzing cancer genomics, autoimmune diseases, or drug responses, OmicsFormer provides the tools you need to extract meaningful insights from complex multi-omics datasets.
Get Started:
Questions? Feedback? I’d love to hear from you! Leave a comment below or reach out via email or GitHub.
Tags: #MachineLearning #DeepLearning #Bioinformatics #MultiOmics #Genomics #Transcriptomics #Proteomics #ComputationalBiology #DataScience #PyTorch #OpenSource #Transformers #PrecisionMedicine #Biomarkers #DrugDiscovery